Pharmacotherapeutic group: antivirals for systemic use, nucleoside and nucleotide reverse transcriptase inhibitors.
ATC code: J05AF10.
PHARMACOLOGY: Pharmacodynamics: Mechanism of action: entecavir, a guanosine nucleoside analogue with activity against HBV polymerase, is efficiently phosphorylated to the active triphosphate (TP) form, which has an intracellular half-life of 15 hours. By competing with the natural substrate deoxyguanosine TP, entecavir-TP functionally inhibits the 3 activities of the viral polymerase: (1) priming of the HBV polymerase, (2) reverse transcription of the negative strand DNA from the pregenomic messenger RNA, and (3) synthesis of the positive strand HBV DNA. The entecavir-TP K
i for HBV DNA polymerase is 0.0012 μM. Entecavir-TP is a weak inhibitor of cellular DNA polymerases α, β, and δ with K
i values of 18 to 40 μM. In addition, high exposures of entecavir had no relevant adverse effects on γ polymerase or mitochondrial DNA synthesis in HepG2 cells (K
i > 160 μM).
Antiviral activity: entecavir inhibited HBV DNA synthesis (50% reduction, EC
50) at a concentration of 0.004 μM in human HepG2 cells transfected with wild-type HBV. The median EC
50 value for entecavir against LVDr HBV (rtL180M and rtM204V) was 0.026 μM (range 0.010-0.059 μM). Recombinant viruses encoding adefovir-resistant substitutions at either rtN236T or rtA181V remained fully susceptible to entecavir.
An analysis of the inhibitory activity of entecavir against a panel of laboratory and clinical HIV-1 isolates using a variety of cells and assay conditions yielded EC
50 values ranging from 0.026 to > 10 μM; the lower EC
50 values were observed when decreased levels of virus were used in the assay.
In cell culture, entecavir selected for an M184I substitution at micromolar concentrations, confirming inhibitory pressure at high entecavir concentrations. HIV variants containing the M184V substitution showed loss of susceptibility to entecavir (see Precautions).
In HBV combination assays in cell culture, abacavir, didanosine, lamivudine, stavudine, tenofovir or zidovudine were not antagonistic to the anti-HBV activity of entecavir over a wide range of concentrations. In HIV antiviral assays, entecavir at micromolar concentrations was not antagonistic to the anti-HIV activity in cell culture of these six NRTIs or emtricitabine.
Resistance in cell culture: relative to wild-type HBV, LVDr viruses containing rtM204V and rtL180M substitutions within the reverse transcriptase exhibit 8-fold decreased susceptibility to entecavir. Incorporation of additional ETVr amino acid changes rtT184, rtS202 or rtM250 decreases entecavir susceptibility in cell culture. Substitutions observed in clinical isolates (rtT184A, C, F, G, I, L, M or S; rtS202 C, G or I; and/or rtM250I, L or V) further decreased entecavir susceptibility. The ETVr substitutions at residues rtT184, rtS202 and rtM250 alone have only a modest effect on entecavir susceptibility, and have not been observed in the absence of LVDr substitutions in more than 1000 patient samples sequenced. Resistance is mediated by reduced inhibitor binding to the altered HBV reverse transcriptase, and resistant HBV exhibits reduced replication capacity in cell culture.
Clinical experience: the demonstration of benefit is based on histological, virological, biochemical, and serological responses after 48 weeks of treatment in active-controlled clinical trials of 1,633 adults with chronic hepatitis B infection, evidence of viral replication and compensated liver disease. The safety and efficacy of entecavir were also evaluated in an active-controlled clinical trial of 191 HBV-infected patients with decompensated liver disease and in a clinical trial of 68 patients co-infected with HBV and HIV.
In studies in patients with compensated liver disease, histological improvement was defined as a ≥ 2-point decrease in Knodell necro-inflammatory score from baseline with no worsening of the Knodell fibrosis score. Responses for patients with baseline Knodell Fibrosis Scores of 4 (cirrhosis) were comparable to overall responses on all efficacy outcome measures (all patients had compensated liver disease). High baseline Knodell necroinflammatory scores (> 10) were associated with greater histological improvement in nucleoside-naive patients. Baseline ALT levels ≥ 2 times ULN and baseline HBV DNA ≤ 9.0 log
10 copies/ml were both associated with higher rates of virologic response (Week 48 HBV DNA < 400 copies/ml) in nucleoside-naive HBeAg-positive patients. Regardless of baseline characteristics, the majority of patients showed histological and virological responses to treatment.
Experience in nucleoside-naive patients with compensated liver disease: Results at 48 weeks of randomised, double blind studies comparing entecavir (ETV) to lamivudine (LVD) in HBeAg positive (022) and HBeAg negative (027) patients are presented in the table. (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Experience in lamivudine-refractory patients with compensated liver disease: In a randomised, double-blind study in HBeAg positive lamivudine-refractory patients (026), with 85% of patients presenting LVDr mutations at baseline, patients receiving lamivudine at study entry either switched to entecavir 1 mg once daily, with neither a washout nor an overlap period (n = 141), or continued on lamivudine 100 mg once daily (n = 145). Results at 48 weeks are presented in the table. (See Table 2.)
Click on icon to see table/diagram/image
Results beyond 48 weeks of treatment: Treatment was discontinued when prespecified response criteria were met either at 48 weeks or during the second year of treatment. Response criteria were HBV virological suppression (HBV DNA < 0.7 MEq/ml by bDNA) and loss of HBeAg (in HBeAg positive patients) or ALT < 1.25 times ULN (in HBeAg negative patients). Patients in response were followed for an additional 24 weeks off-treatment. Patients who met virologic but not serologic or biochemical response criteria continued blinded treatment. Patients who did not have a virologic response were offered alternative treatment.
Nucleoside-naive: HBeAg positive (study 022): treatment with entecavir for up to 96 weeks (n = 354) resulted in cumulative response rates of 80% for HBV DNA < 300 copies/ml by PCR, 87% for ALT normalisation, 31% for HBeAg seroconversion and 2% for HBsAg seroconversion (5% for HBsAg loss). For lamivudine (n = 355), cumulative response rates were 39% for HBV DNA < 300 copies/ml by PCR, 79% for ALT normalisation, 26% for HBeAg seroconversion, and 2% for HBsAg seroconversion (3% for HBsAg loss).
At end of dosing, among patients who continued treatment beyond 52 weeks (median of 96 weeks), 81% of 243 entecavir-treated and 39% of 164 lamivudine-treated patients had HBV DNA < 300 copies/ml by PCR while ALT normalisation (≤ 1 times ULN) occurred in 79% of entecavir-treated and 68% of lamivudine-treated patients.
HBeAg negative (study 027): treatment with entecavir up to 96 weeks (n = 325) resulted in cumulative response rates of 94% for HBV DNA < 300 copies/ml by PCR and 89% for ALT normalisation versus 77% for HBV DNA < 300 copies/ml by PCR and 84% for ALT normalisation for lamivudine-treated patients (n = 313).
For 26 entecavir-treated and 28 lamivudine-treated patients who continued treatment beyond 52 weeks (median 96 weeks), 96% of entecavir-treated and 64% of lamivudine-treated patients had HBV DNA < 300 copies/ml by PCR at end of dosing. ALT normalisation (≤ 1 times ULN) occurred in 27% of entecavir-treated and 21% of lamivudine-treated patients at end of dosing.
For patients who met protocol-defined response criteria, response was sustained throughout the 24-week post-treatment follow-up in 75% (83/111) of entecavir responders vs 73% (68/93) for lamivudine responders in study 022 and 46% (131/286) of entecavir responders vs 31% (79/253) for lamivudine responders in study 027. By 48 weeks of post-treatment follow-up, a substantial number of HBeAg negative patients lost response.
Liver biopsy results: 57 patients from the pivotal nucleoside-naive studies 022 (HBeAg positive) and 027 (HBeAg negative) who enrolled in a long-term rollover study were evaluated for long-term liver histology outcomes. The entecavir dosage was 0.5 mg daily in the pivotal studies (mean exposure 85 weeks) and 1 mg daily in the rollover study (mean exposure 177 weeks), and 51 patients in the rollover study initially also received lamivudine (median duration 29 weeks). Of these patients, 55/57 (96%) had histological improvement as previously defined (see previous text), and 50/57 (88%) had a ≥ 1-point decrease in Ishak fibrosis score. For patients with baseline Ishak fibrosis score ≥ 2, 25/43 (58%) had a ≥ 2-point decrease. All (10/10) patients with advanced fibrosis or cirrhosis at baseline (Ishak fibrosis score of 4, 5 or 6) had a ≥ 1 point decrease (median decrease from baseline was 1.5 points). At the time of the long-term biopsy, all patients had HBV DNA < 300 copies/ml and 49/57 (86%) had serum ALT ≤ 1 times ULN. All 57 patients remained positive for HBsAg.
Lamivudine-refractory: HBeAg positive (study 026): treatment with entecavir for up to 96 weeks (n = 141) resulted in cumulative response rates of 30% for HBV DNA < 300 copies/ml by PCR, 85% for ALT normalisation and 17% for HBeAg seroconversion.
For the 77 patients who continued entecavir treatment beyond 52 weeks (median 96 weeks), 40% of patients had HBV DNA < 300 copies/ml by PCR and 81% had ALT normalisation (≤ 1 times ULN) at end of dosing.
Age/gender: There was no apparent difference in efficacy for entecavir based on gender (≈ 25% women in the clinical trials) or age (≈ 5% of patients > 65 years of age).
Long-Term Follow-Up Study: Study 080 was a randomized, observational open-label Phase 4 study to assess long-term risks of entecavir treatment (ETV, n=6,216) or other standard of care HBV nucleoside (acid) treatment (non-ETV) (n=6,162) for up to 10 years in subjects with chronic HBV (CHB) infection. The principal clinical outcome events assessed in the study were overall malignant neoplasms (composite event of HCC and non-HCC malignant neoplasms), liver related HBV disease progression, non-HCC malignant neoplasms, HCC, and deaths, including liver related deaths. In this study, ETV was not associated with an increased risk of malignant neoplasms compared to use of non-ETV, as assessed by either the composite endpoint of overall malignant neoplasms (ETV n=331, non-ETV n=337; HR=0.93 [0.8-1.1]), or the individual endpoint of non-HCC malignant neoplasm (ETV n=95, non-ETV n=81; HR=1.1 [0.82-1.5]). The reported events for liver-related HBV disease progression and HCC were comparable in both ETV and non-ETV groups. The most commonly reported malignancy in both ETV and non-ETV groups was HCC followed by gastrointestinal malignancies.
Special populations: Patients with decompensated liver disease: in study 048, 191 patients with HBeAg positive or negative chronic HBV infection and evidence of hepatic decompensation, defined as a CTP score of 7 or higher, received entecavir 1 mg once daily or adefovir dipivoxil 10 mg once daily. Patients were either HBV-treatment-naïve or pretreated (excluding pretreatment with entecavir, adefovir dipivoxil, or tenofovir disoproxil fumarate). At baseline, patients had a mean CTP score of 8.59 and 26% of patients were CTP class C. The mean baseline Model for End Stage Liver Disease (MELD) score was 16.23. Mean serum HBV DNA by PCR was 7.83 log
10 copies/ml and mean serum ALT was 100 U/l; 54% of patients were HBeAg positive, and 35% of patients had LVDr substitutions at baseline. Entecavir was superior to adefovir dipivoxil on the primary efficacy endpoint of mean change from baseline in serum HBV DNA by PCR at week 24. Results for selected study endpoints at weeks 24 and 48 are shown in the table. (See Table 3.)
Click on icon to see table/diagram/image
The time to onset of HCC or death (whichever occurred first) was comparable in the two treatment groups; on-study cumulative death rates were 23% (23/102) and 33% (29/89) for patients treated with entecavir and adefovir dipivoxil, respectively, and on-study cumulative rates of HCC were 12% (12/102) and 20% (18/89) for entecavir and adefovir dipivoxil, respectively. For patients with LVDr substitutions at baseline, the percentage of patients with HBV DNA <300 copies/ml was 44% for entecavir and 20% for adefovir at week 24 and 50% for entecavir and 17% for adefovir at week 48.
HIV/HBV co-infected patients receiving concomitant HAART: study 038 included 67 HBeAg positive and 1 HBeAg negative patients co-infected with HIV. Patients had stable controlled HIV (HIV RNA < 400 copies/ml) with recurrence of HBV viraemia on a lamivudine-containing HAART regimen. HAART regimens did not include emtricitabine or tenofovir disoproxil fumarate. At baseline entecavir-treated patients had a median duration of prior lamivudine therapy of 4.8 years and median CD4 count of 494 cells/mm
3 (with only 5 subjects having CD4 count < 200 cells/mm
3). Patients continued their lamivudine-regimen and were assigned to add either entecavir 1 mg once daily (n = 51) or placebo (n = 17) for 24 weeks followed by an additional 24 weeks where all received entecavir. At 24 weeks the reduction in HBV viral load was significantly greater with entecavir (-3.65 vs an increase of 0.11 log
10 copies/ml). For patients originally assigned to entecavir treatment, the reduction in HBV DNA at 48 weeks was -4.20 log
10 copies/ml, ALT normalisation had occurred in 37% of patients with abnormal baseline ALT and none achieved HBeAg seroconversion.
HIV/HBV co-infected patients not receiving concomitant HAART: entecavir has not been evaluated in HIV/HBV co-infected patients not concurrently receiving effective HIV treatment. Reductions in HIV RNA have been reported in HIV/HBV co-infected patients receiving entecavir monotherapy without HAART. In some cases, selection of HIV variant M184V has been observed, which has implications for the selection of HAART regimens that the patient may take in the future. Therefore, entecavir should not be used in this setting due to the potential for development of HIV resistance (see Precautions).
Liver transplant recipients: the safety and efficacy of entecavir 1 mg once daily were assessed in a single-arm study in 65 patients who received a liver transplant for complications of chronic HBV infection and had HBV DNA <172 IU/ml (approximately 1000 copies/ml) at the time of transplant. The study population was 82% male, 39% Caucasian, and 37% Asian, with a mean age of 49 years; 89% of patients had HBeAg-negative disease at the time of transplant. Of the 61 patients who were evaluable for efficacy (received entecavir for at least 1 month), 60 also received hepatitis B immune globulin (HBIg) as part of the post-transplant prophylaxis regimen. Of these 60 patients, 49 received more than 6 months of HBIg therapy. At Week 72 post-transplant, none of 55 observed cases had virologic recurrence of HBV [defined as HBV DNA ≥50 IU/ml (approximately 300 copies/ml)], and there was no reported virologic recurrence at time of censoring for the remaining 6 patients. All 61 patients had HBsAg loss post-transplantation, and 2 of these later became HBsAg positive despite maintaining undetectable HBV DNA (<6 IU/ml). The frequency and nature of adverse events in this study were consistent with those expected in patients who have received a liver transplant and the known safety profile of entecavir.
Paediatric population: Study 189 is an ongoing study of the efficacy and safety of entecavir among 180 nucleoside-treatment-naïve children and adolescents from 2 to < 18 years of age with HBeAg-positive chronic hepatitis B infection, compensated liver disease, and elevated ALT. Patients were randomized (2:1) to receive blinded treatment with entecavir 0.015 mg/kg up to 0.5 mg/day (N = 120) or placebo (N = 60). The randomization was stratified by age group (2 to 6 years; > 6 to 12 years; and > 12 to < 18 years). Baseline demographics and HBV disease characteristics were comparable between the 2 treatment arms and across age cohorts. At study entry, the mean HBV DNA was 8.1 log
10 IU/ml and mean ALT was 103 U/l across the study population. Results for the main efficacy endpoints at Week 48 and Week 96 are presented in the table as follows. (See Table 4.)
Click on icon to see table/diagram/image
The paediatric resistance assessment is based on data from nucleoside-treatment-naive paediatric patients with HBeAg-positive chronic HBV infection in two ongoing clinical trials (028 and 189). The two trials provide resistance data in 183 patients treated and monitored in Year 1 and 180 patients treated and monitored in Year 2. Genotypic evaluations were performed for all patients with available samples who had virologic breakthrough through Week 96 or HBV DNA ≥ 50 IU/ml at Week 48 or Week 96. During Year 2, genotypic resistance to ETV was detected in 2 patients (1.1% cumulative probability of resistance through Year 2).
Clinical resistance in Adults: patients in clinical trials initially treated with entecavir 0.5 mg (nucleoside-naive) or 1.0 mg (lamivudine-refractory) and with an on-therapy PCR HBV DNA measurement at or after Week 24 were monitored for resistance.
Through Week 240 in nucleoside-naive studies, genotypic evidence of ETVr substitutions at rtT184, rtS202, or rtM250 was identified in 3 patients treated with entecavir, 2 of whom experienced virologic breakthrough (see Table). These substitutions were observed only in the presence of LVDr substitutions (rtM204V and rtL180M). (See Table 5.)
Click on icon to see table/diagram/image
ETVr substitutions (in addition to LVDr substitutions rtM204V/I ± rtL180M) were observed at baseline in isolates from 10/187 (5%) lamivudine-refractory patients treated with entecavir and monitored for resistance, indicating that prior lamivudine treatment can select these resistance substitutions and that they can exist at a low frequency before entecavir treatment. Through Week 240, 3 of the 10 patients experienced virologic breakthrough (≥ 1 log
10 increase above nadir). Emerging entecavir resistance in lamivudine-refractory studies through Week 240 is summarized in the table. (See Table 6.)
Click on icon to see table/diagram/image
Among lamivudine-refractory patients with baseline HBV DNA < 10
7 log
10 copies/ml, 64% (9/14) achieved HBV DNA < 300 copies/ml at Week 48. These 14 patients had a lower rate of genotypic entecavir resistance (cumulative probability 18.8% through 5 years of follow-up) than the overall study population (see Table). Also, lamivudine-refractory patients who achieved HBV DNA < 10
4 log
10 copies/ml by PCR at Week 24 had a lower rate of resistance than those who did not (5-year cumulative probability 17.6% [n=50] versus 60.5% [n=135], respectively).
Integrated Analysis of Phase 2 and 3 Clinical Studies: In a post-approval integrated analysis of entecavir resistance data from 17 Phase 2 and 3 clinical studies, an emergent entecavir resistance-associated substitution rtA181C was detected in 5 out of 1461 subjects during treatment with entecavir. This substitution was detected only in the presence of lamivudine resistance-associated substitutions rtL180M plus rtM204V.
Pharmacokinetics: Absorption: entecavir is rapidly absorbed with peak plasma concentrations occurring between 0.5-1.5 hours. The absolute bioavailability has not been determined. Based on urinary excretion of unchanged drug, the bioavailability has been estimated to be at least 70%. There is a dose-proportionate increase in C
max and AUC values following multiple doses ranging from 0.1-1 mg. Steady-state is achieved between 6-10 days after once daily dosing with ≈ 2 times accumulation. C
max and C
min at steady-state are 4.2 and 0.3 ng/ml, respectively, for a dose of 0.5 mg, and 8.2 and 0.5 ng/ml, respectively, for 1 mg. The tablet and oral solution were bioequivalent in healthy subjects; therefore, both forms may be used interchangeably.
Administration of 0.5 mg entecavir with a standard high-fat meal (945 kcal, 54.6 g fat) or a light meal (379 kcal, 8.2 g fat) resulted in a minimal delay in absorption (1-1.5 hour fed vs. 0.75 hour fasted), a decrease in C
max of 44-46%, and a decrease in AUC of 18-20%. The lower C
max and AUC when taken with food is not considered to be of clinical relevance in nucleoside-naive patients but could affect efficacy in lamivudine-refractory patients (see Dosage & Administration).
Distribution: the estimated volume of distribution for entecavir is in excess of total body water. Protein binding to human serum protein
in vitro is ≈ 13%.
Biotransformation: entecavir is not a substrate, inhibitor or inducer of the CYP450 enzyme system. Following administration of
14C-entecavir, no oxidative or acetylated metabolites and minor amounts of the phase II metabolites, glucuronide and sulfate conjugates, were observed.
Elimination: entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at steady-state of about 75% of the dose. Renal clearance is independent of dose and ranges between 360-471 ml/min suggesting that entecavir undergoes both glomerular filtration and net tubular secretion. After reaching peak levels, entecavir plasma concentrations decreased in a bi-exponential manner with a terminal elimination half-life of ≈ 128-149 hours. The observed drug accumulation index is ≈ 2 times with once daily dosing, suggesting an effective accumulation half-life of about 24 hours.
Hepatic impairment: pharmacokinetic parameters in patients with moderate or severe hepatic impairment were similar to those in patients with normal hepatic function.
Renal impairment: entecavir clearance decreases with decreasing creatinine clearance. A 4 hour period of haemodialysis removed ≈ 13% of the dose, and 0.3% was removed by CAPD. The pharmacokinetics of entecavir following a single 1 mg dose in patients (without chronic hepatitis B infection) are shown in the table as follows: (See Table 7.)
Click on icon to see table/diagram/image
Post-Liver transplant: entecavir exposure in HBV-infected liver transplant recipients on a stable dose of cyclosporine A or tacrolimus (n = 9) was ≈ 2 times the exposure in healthy subjects with normal renal function. Altered renal function contributed to the increase in entecavir exposure in these patients (see Precautions).
Gender: AUC was 14% higher in women than in men, due to differences in renal function and weight. After adjusting for differences in creatinine clearance and body weight there was no difference in exposure between male and female subjects.
Elderly: the effect of age on the pharmacokinetics of entecavir was evaluated comparing elderly subjects in the age range 65-83 years (mean age females 69 years, males 74 years) with young subjects in the age range 20-40 years (mean age females 29 years, males 25 years). AUC was 29% higher in elderly than in young subjects, mainly due to differences in renal function and weight. After adjusting for differences in creatinine clearance and body weight, elderly subjects had a 12.5% higher AUC than young subjects. The population pharmacokinetic analysis covering patients in the age range 16-75 years did not identify age as significantly influencing entecavir pharmacokinetics.
Race: the population pharmacokinetic analysis did not identify race as significantly influencing entecavir pharmacokinetics. However, conclusions can only be drawn for the Caucasian and Asian groups as there were too few subjects in the other categories.
Paediatric population: the steady-state pharmacokinetics of entecavir were evaluated (study 028) in 24 nucleoside naïve and 19 lamivudine-experienced HBeAg-positive paediatric subjects from 2 to < 18 years of age with compensated liver disease. Entecavir exposure among nucleoside naïve subjects receiving once daily doses of entecavir 0.015 mg/kg up to a maximum dose of 0.5 mg was similar to the exposure achieved in adults receiving once daily doses of 0.5 mg. The C
max, AUC(0-24), and C
min for these subjects was 6.31 ng/ml, 18.33 ng h/ml, and 0.28 ng/ml, respectively.
Toxicology: Preclinical safety data: In repeat-dose toxicology studies in dogs, reversible perivascular inflammation was observed in the central nervous system, for which no-effect doses corresponded to exposures 19 and 10 times those in humans (at 0.5 and 1 mg respectively). This finding was not observed in repeat-dose studies in other species, including monkeys administered entecavir daily for 1 year at exposures ≥ 100 times those in humans.
In reproductive toxicology studies in which animals were administered entecavir for up to 4 weeks, no evidence of impaired fertility was seen in male or female rats at high exposures. Testicular changes (seminiferous tubular degeneration) were evident in repeat-dose toxicology studies in rodents and dogs at exposures ≥ 26 times those in humans. No testicular changes were evident in a 1-year study in monkeys.
In pregnant rats and rabbits administered entecavir, no effect levels for embryotoxicity and maternal toxicity corresponded to exposures ≥ 21 times those in humans. In rats, maternal toxicity, embryo-foetal toxicity (resorptions), lower foetal body weights, tail and vertebral malformations, reduced ossification (vertebrae, sternebrae, and phalanges), and extra lumbar vertebrae and ribs were observed at high exposures. In rabbits, embryo-foetal toxicity (resorptions), reduced ossification (hyoid), and an increased incidence of 13th rib were observed at high exposures. In a peri-postnatal study in rats, no adverse effects on offspring were observed. In a separate study wherein entecavir was administered to pregnant lactating rats at 10 mg/kg, both foetal exposure to entecavir and secretion of entecavir into milk were demonstrated. In juvenile rats administered entecavir from postnatal days 4 to 80, a moderately reduced acoustic startle response was noted during the recovery period (postnatal days 110 to 114) but not during the dosing period at AUC values ≥ 92 times those in humans at the 0.5 mg dose or paediatric equivalent dose. Given the exposure margin, this finding is considered of unlikely clinical significance.
No evidence of genotoxicity was observed in an Ames microbial mutagenicity assay, a mammalian-cell gene mutation assay, and a transformation assay with Syrian hamster embryo cells. A micronucleus study and a DNA repair study in rats were also negative. Entecavir was clastogenic to human lymphocyte cultures at concentrations substantially higher than those achieved clinically.
Two-year carcinogenicity studies: in male mice, increases in the incidences of lung tumours were observed at exposures ≥ 4 and ≥ 2 times that in humans at 0.5 mg and 1 mg respectively. Tumour development was preceded by pneumocyte proliferation in the lung which was not observed in rats, dogs, or monkeys, indicating that a key event in lung tumour development observed in mice likely was species-specific. Increased incidences of other tumours including brain gliomas in male and female rats, liver carcinomas in male mice, benign vascular tumours in female mice, and liver adenomas and carcinomas in female rats were seen only at high lifetime exposures. However, the no effect levels could not be precisely established. The predictivity of the findings for humans is not known. For clinical data, see PHARMACOLOGY: Pharmacodynamics as previously mentioned.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image Click on icon to see table/diagram/image
Click on icon to see table/diagram/image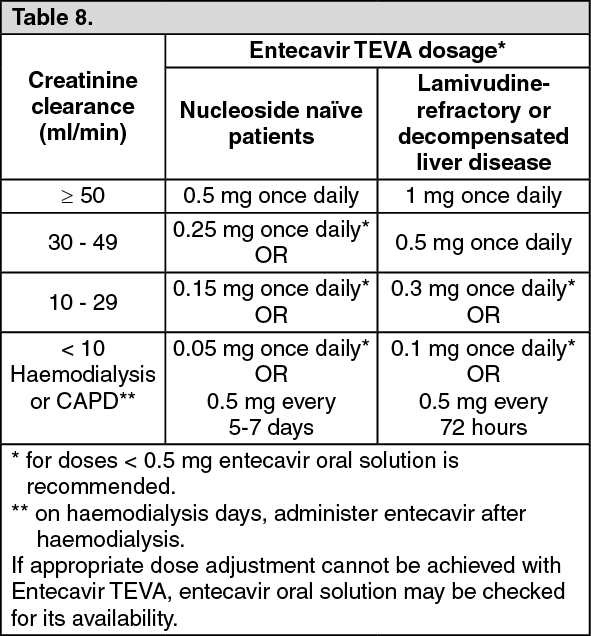 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image

 Sign Out
Sign Out
